Reactions¶
In this system, the only actual reaction is between a C1 of one 4-methylstyrene monomer and the C2 of another:
- name: emb1_1
stage: cure
reactants:
1: EMB
2: EMB
product: EMB~C1-C2~EMB
probability: 1.0
atoms:
A:
reactant: 1
resid: 1
atom: C1
z: 1
B:
reactant: 2
resid: 1
atom: C2
z: 1
bonds:
- atoms:
- A
- B
order: 1
Naturally, this generates a dimer:
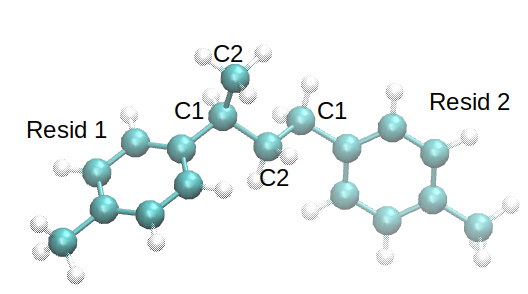
Notice that atom C1 of resid 1 has “attacked” C2 of resid 2 to form the bond.
Now, consider the new bonded interactions that spring from the creation of this one bond. Clearly, it is one side of a bunch of new angles, each of which is defined by one other atom on either resid 1 or 2. It is also the central bond of many new proper dihedrals, each of which involves one neighbor atom each on resid 1 and another on resid 2. Clearly, the dimer is sufficient to permit parameterization to yield these interactions.
However, that is not all. The new bond is also one of the outer bonds of many dihedrals. For example, the 1-C1–2-C2 bond is part of a torsion in which the 1-C2–1-C1 bond is the central bond, meaning each other atom on C2 of resid 1 defines a unique proper dihedral. In this dimer, all those atoms are hydrogens. However, what if C2 of resid 1 is attacked by C1 of a third monomer? Then one of these dihedrals is actually a C-C-C-C dihedral, not an H-C-C-C dihedral. This means that, generally, for systems with monomers that react via double-bond opening, we need oligomeric templats that involve 3 and 4 monomers to have a full set of templates.
HTPolyNet builds these template molecules automatically. The central concept it uses is termed a “chain”. By definition, a chain is any contiguous sequence of reactive atoms. An unreacted EMB molecule has a chain of length two composed of atoms C1 and C2. If two EMB molecules react to form a dimer, their two intramolecular “chains” merge to form a single chain C1-C2-C1-C2 (where the first two atoms are in one monomer and the second two are in the other). Given the monomer structures and reaction definitions, HTPolyNet first determines if chains can exist at all, and if so, it performs a so-called “chain-expansion” of all reactions. That is, since the base reactions listed explicitly in a configuration file typically join two monomers with a bond, if that bond can be in a chain, then HTPolyNet automatically generates all the other possible reactions in which that particular monomer-monomer linkage is possible for products that contain three and four monomers. This guarantees that all possible bonded topologies are represented among templates out to sufficient numbers of atoms to permit mapping of atom types and charges back onto the actual system.
In this case, there are three additional reactions that are generated by chain expansion:
The reaction in which a C1 of EMB attacks the C2 of the first EMB in an EMB-EMB dimer;
The reaction in which a C1 of the second EMB of an EMB-EMB dimer attacks the C2 of an EMB monomer; and
The reaction in which a C1 of the second EMB of an EMB-EMB dimer attacks the C2 of the first EMB of another EMB-EMB dimer.
Each of these reactions generates a unique template molecule; the first two generate EMB trimers and the third generates an EMB quadrimer. Although the two trimers are identical molecules, the particular bond that forms to generate each trimer is different for each one, and for now, HTPolyNet treats them as different molecules (this is admittedly inefficient but it works).
Examining detailed debugging logs, we can see HTPolyNet in action generating these three new reactions:
monomer atom EMB_C1 will attack dimer atom EMB1_1[EMB1_C2] -> EMB~C1=C2~EMB1_1:
Reaction "emb~c1=c2~emb~c1-c2~emb"
reactant 1: EMB
reactant 2: EMB1_1
product EMB~C1=C2~EMB~C1-C2~EMB
atom A: {'reactant': 1, 'resid': 1, 'atom': 'C1', 'z': 1}
atom B: {'reactant': 2, 'resid': 1, 'atom': 'C2', 'z': 1}
bond {'atoms': ['A', 'B'], 'order': 1}
dimer atom EMB~C1-C2~EMB[EMB2_C1] will attack monomer atom EMB_C2-> EMB~C1-C2~EMB~C1=C2~EMB:
Reaction "emb1_1~c1=c2~emb"
reactant 1: EMB~C1-C2~EMB
reactant 2: EMB
product EMB~C1-C2~EMB~C1=C2~EMB
atom A: {'reactant': 1, 'resid': 2, 'atom': 'C1', 'z': 1}
atom B: {'reactant': 2, 'resid': 1, 'atom': 'C2', 'z': 1}
bond {'atoms': ['A', 'B'], 'order': 1}
dimer atom EMB~C1-C2~EMB[EMB2_C1] will attack dimer atom EMB~C1-C2~EMB[EMB1_C2] -> EMB~C1-C2~EMB~C1=C2~EMB~C1-C2~EMB:
Reaction "emb1_1~c1=c2~emb1_1"
reactant 1: EMB~C1-C2~EMB
reactant 2: EMB~C1-C2~EMB
product EMB~C1-C2~EMB~C1=C2~EMB~C1-C2~EMB
atom A: {'reactant': 1, 'resid': 2, 'atom': 'C1', 'z': 1}
atom B: {'reactant': 2, 'resid': 1, 'atom': 'C2', 'z': 1}
bond {'atoms': ['A', 'B'], 'order': 1}
Finally, we can choose to revert any fully unreacted monomers back to true double bonds (i.e., “cap” the bonds):
- name: embCC
stage: cap
reactants:
1: EMB
product: EMBCC
probability: 1.0
atoms:
A:
reactant: 1
resid: 1
atom: C1
z: 1
B:
reactant: 1
resid: 1
atom: C2
z: 1
bonds:
- atoms:
- A
- B
order: 2
The next thing we consider is the configuration file necessary to direct the build.