Monomer: 4-methylstyrene¶
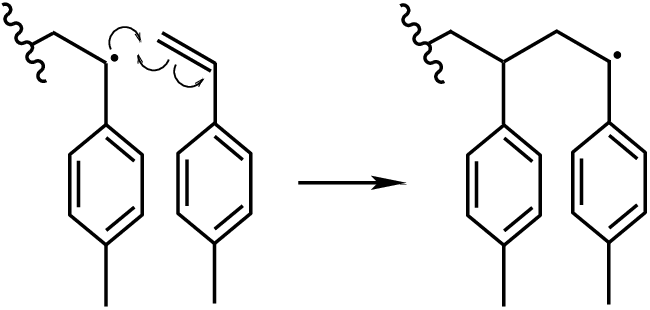
Fig. 6 Polymerization of methyl styrene.¶
4-methylstyrene is a common monomer in the manufacture of polyesters. Its most important feature from the standpoint of polymerization is the carbon-carbon double bond, which opens to form a single bond and a radical if attacked by another radical (Fig. 6). Note from Fig. 6 that the radical lives on the interior carbon and attacks a terminal carbon of another unreacted monomer to create a bond. Clearly, then, HTPolyNet must be able to distinguish between these two carbon atoms. To see how we do that, let’s turn to a way to build the mol2 file that describes the monomer structure and topology.
As described in the user guide, HTPolyNet uses the concepts of “valence conservation” and “sacrificial hydrogens”: any two atoms designated as forming a bond must each sacrifice one H atom to make the bond. The form of 4-methylstyrene we will actually use to build our system will be 1-ethyl-4-methylbenzene. We can easily generate a mol2 file for 1-ethyl-4-methylbenzene using OpenBabel (or any of a variety of molecular builders):
$ echo "C1=CC(C)=CC=C1CC" | obabel -ismi --gen3d -h -omol2 > EMB-raw.mol2
Now, let’s have a look at this file (your coordinates may be different):
@<TRIPOS>MOLECULE
EMB
21 21 0 0 0
SMALL
GASTEIGER
@<TRIPOS>ATOM
1 C -0.7183 1.1964 -0.0469 C.ar 1 EMB -0.0583
2 C 0.6786 1.2489 -0.0406 C.ar 1 EMB -0.0586
3 C 1.4326 0.0702 -0.0692 C.ar 1 EMB -0.0504
4 C 2.9313 0.1119 -0.1179 C.3 1 EMB -0.0397
5 C 0.7663 -1.1602 -0.0643 C.ar 1 EMB -0.0586
6 C -0.6303 -1.2130 -0.0702 C.ar 1 EMB -0.0583
7 C -1.3840 -0.0340 -0.0882 C.ar 1 EMB -0.0476
8 C -2.8901 -0.0895 -0.1745 C.3 1 EMB -0.0305
9 C -3.5635 -0.1866 1.1846 C.3 1 EMB -0.0613
10 H -1.2827 2.1250 -0.0377 H 1 EMB 0.0620
11 H 1.1716 2.2188 -0.0290 H 1 EMB 0.0620
12 H 3.3028 1.1393 -0.1869 H 1 EMB 0.0278
13 H 3.2926 -0.4300 -0.9987 H 1 EMB 0.0278
14 H 3.3531 -0.3437 0.7826 H 1 EMB 0.0278
15 H 1.3333 -2.0885 -0.0716 H 1 EMB 0.0620
16 H -1.1240 -2.1816 -0.0811 H 1 EMB 0.0620
17 H -3.1842 -0.9482 -0.7919 H 1 EMB 0.0311
18 H -3.2577 0.7973 -0.7038 H 1 EMB 0.0311
19 H -3.2530 -1.0884 1.7216 H 1 EMB 0.0233
20 H -4.6512 -0.2259 1.0596 H 1 EMB 0.0233
21 H -3.3263 0.6802 1.8094 H 1 EMB 0.0233
@<TRIPOS>BOND
1 1 2 ar
2 2 3 ar
3 3 4 1
4 3 5 ar
5 5 6 ar
6 6 7 ar
7 1 7 ar
8 7 8 1
9 8 9 1
10 1 10 1
11 2 11 1
12 4 12 1
13 4 13 1
14 4 14 1
15 5 15 1
16 6 16 1
17 8 17 1
18 8 18 1
19 9 19 1
20 9 20 1
21 9 21 1
Notice how the atom names (second column in the @<TRIPOS>ATOM section) are not unique? This is potentially a problem, since HTPolyNet always refers to particular atoms by virtue of their “residue name” and “name”. (There is only one residue here, called EMB.) Let’s call the radical-bearing carbon C1 and the methyl carbon C2. To figure out which atoms these are in the mol2 file, we can interrogate the structure in VMD (or any other suitable visualization software):
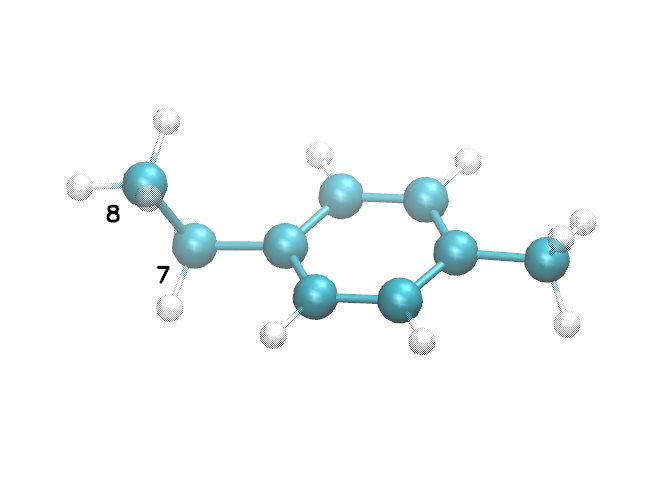
The black numbers shown here indicate internal atom indexes in VMD, and VMD starts counting at zero. Mol2 and Gromacs start counting at 1, so these atoms’ indexes are one more than what is shown here. We see the methylene carbon is index 7 in VMD, so it is index 8 in the mol2 file; likewise, the methyl carbon is index 8 in VMD and so index 9 in the mol2 file. Let’s use this information along to force obabel to give us a ready-to-use mol2 file:
$ echo "C1=CC(C)=CC=C1CC" | \
obabel -ismi --gen3d -h -omol2 --title "EMB" | \
sed s/" 8 C "/" 8 C1"/ | \
sed s/" 9 C "/" 9 C2"/ | \
sed s/"UNL1"/"EMB "/ > EMB.mol2
Let’s look at the file EMB.mol2 that results from the command above:
@<TRIPOS>MOLECULE
EMB
21 21 0 0 0
SMALL
GASTEIGER
@<TRIPOS>ATOM
1 C -0.7183 1.1964 -0.0469 C.ar 1 EMB -0.0583
2 C 0.6786 1.2489 -0.0406 C.ar 1 EMB -0.0586
3 C 1.4326 0.0702 -0.0692 C.ar 1 EMB -0.0504
4 C 2.9313 0.1119 -0.1179 C.3 1 EMB -0.0397
5 C 0.7663 -1.1602 -0.0643 C.ar 1 EMB -0.0586
6 C -0.6303 -1.2130 -0.0702 C.ar 1 EMB -0.0583
7 C -1.3840 -0.0340 -0.0882 C.ar 1 EMB -0.0476
8 C1 -2.8901 -0.0895 -0.1745 C.3 1 EMB -0.0305
9 C2 -3.5635 -0.1866 1.1846 C.3 1 EMB -0.0613
10 H -1.2827 2.1250 -0.0377 H 1 EMB 0.0620
11 H 1.1716 2.2188 -0.0290 H 1 EMB 0.0620
12 H 3.3028 1.1393 -0.1869 H 1 EMB 0.0278
13 H 3.2926 -0.4300 -0.9987 H 1 EMB 0.0278
14 H 3.3531 -0.3437 0.7826 H 1 EMB 0.0278
15 H 1.3333 -2.0885 -0.0716 H 1 EMB 0.0620
16 H -1.1240 -2.1816 -0.0811 H 1 EMB 0.0620
17 H -3.1842 -0.9482 -0.7919 H 1 EMB 0.0311
18 H -3.2577 0.7973 -0.7038 H 1 EMB 0.0311
19 H -3.2530 -1.0884 1.7216 H 1 EMB 0.0233
20 H -4.6512 -0.2259 1.0596 H 1 EMB 0.0233
21 H -3.3263 0.6802 1.8094 H 1 EMB 0.0233
@<TRIPOS>BOND
1 1 2 ar
2 2 3 ar
3 3 4 1
4 3 5 ar
5 5 6 ar
6 6 7 ar
7 1 7 ar
8 7 8 1
9 8 9 1
10 1 10 1
11 2 11 1
12 4 12 1
13 4 13 1
14 4 14 1
15 5 15 1
16 6 16 1
17 8 17 1
18 8 18 1
19 9 19 1
20 9 20 1
21 9 21 1
You can see how atoms 8 and 9 (mol2 indexes) are now named C1 and C2, respectively. The command above is embedded in the run.sh script that comes with this example.
The next thing we consider is how to create the reaction dictionaries necessary to describe the polymerization chemistry.