Monomers¶
DGEBA¶
Bisphenol A diglycidyl ether, which we refer to as DGEBA for historical reasons, is an epoxidized form of bisphenol A (BPA). Here we’ll consider how to build the input .mol2 file for DGEBA. It is quite easy to generate a 3D structure from a SMILES representation. The canonical SMILES string for DGEBA is:
CC(C)(C1=CC=C(C=C1)OCC2CO2)C3=CC=C(C=C3)OCC4CO4
However, as described in the user guide, HTPolyNet uses the concept of “sacrificial hydrogens”: any two atoms designated as forming a bond must each sacrifice one H atom to make the bond. Epoxy groups react with amines via hydrogen atom transfer from the amine to the oxirane oxygen, generating a C-N bond and a pendant OH group one carbon atom removed from the C-N bond. So the N sacrificed but the C did not; it “sacrificed” its bond to the O atom of the oxirane. To use DGEBA as a reactive monomer, we therefore must convert it to a form in which the two oxiranes are hydrogenated, yielding a terminal methyl group and a pendant OH one atom removed from the methyl. This is easily done by altering the C2CO2 and C4CO4 epoxirane cycles in the original SMILES string to C(O)C methyl hydroxymethyl groups, yielding:
CC(C)(C1=CC=C(C=C1)OCC(O)C)C3=CC=C(C=C3)OCC(O)C
Using OpenBabel (or any of a variety of molecular builders), we can generate a baseline 3D structure of DGEBA in .mol2 format at the command-line, referring to it as “DGE” (three-letter monomer names are customary, though not required; they need only be unique identifiers):
$ echo "CC(C)(C1=CC=C(C=C1)OCC(O)C)C3=CC=C(C=C3)OCC(O)C" | \
obabel -ismi -h --gen3d -omol2 --title "DGE" | \
sed s/"UNL1 "/"DGE "/ > DGE-raw.mol2"
Note that we have used sed to change the generic residue name UNL1 provided by obabel to our desired name DGE. At this point, although this is a valid *.mol2 file, it is not yet ready for use by HTPolyNet. It needs two fixes:
Reactive atoms must be uniquely named; and
Any chiral carbons must also be uniquely named.
To understand how to make these fixes, we should visualize the molecule:
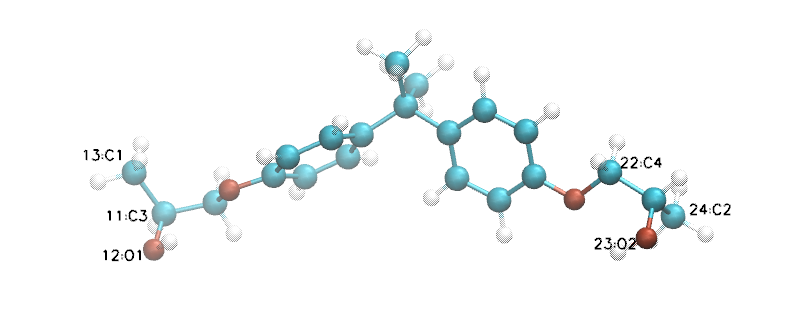
This image was made with VMD, and you can see that four carbons and two oxygens are labelled. The numbers refer to internal atom indices assigned by VMD, which begins counting at zero. These correspond to the atom indices in the *.mol2 file, which begins counting at 1. So the atoms labelled 13 and 24 are atoms 14 and 25 in DGE.mol2; these are the two “reactive” carbons because each can bond to an N of an amine. Furthermore, since oxirane opening usually generates a chiral carbon, we indeed see that the atoms labelled 11 and 22 are both chiral centers, and both in S; these of course are atoms with the indicies 12 and 23 in DGE.mol2. Finally, since we will ultimately want to convert any unreacted epoxies back into oxirane rings, we need to specify the relevant oxygen atoms; these are atom with VMD-indices 12 and 23, which are 13 and 24 in DGE.mol2 file.
Let’s edit DGE.mol2 to name the two reactive atoms C1 and C2, the two chiral atoms at C3 and C4, and the two oxirane oxygens as O1 and O2:
$ cat DGE-raw.mol2 | sed s/"14 C "/"14 C1"/ | \
sed s/"25 C "/"25 C2"/ | \
sed s/"12 C "/"12 C3"/ | \
sed s/"23 C "/"23 C4"/ | \
sed s/"13 O "/"13 O1"/ | \
sed s/"24 O "/"24 O2"/ > DGE.mol2
Note that in the sed substitution directives, we have preserved the number of characters substituted to keep the column spacing in the *.mol2 file from changing.
Now, let’s take a look at DGE.mol2:
@<TRIPOS>MOLECULE
DGE
53 54 0 0 0
SMALL
GASTEIGER
@<TRIPOS>ATOM
1 C 0.9601 0.0682 0.1490 C.3 1 DGE -0.0517
2 C 2.5158 0.0610 0.0695 C.3 1 DGE 0.0151
3 C 2.9888 0.3744 1.5227 C.3 1 DGE -0.0517
4 C 3.0435 -1.3437 -0.3149 C.ar 1 DGE -0.0372
5 C 2.2654 -2.2043 -1.1002 C.ar 1 DGE -0.0543
6 C 2.6154 -3.5414 -1.2810 C.ar 1 DGE -0.0197
7 C 3.7559 -4.0699 -0.6824 C.ar 1 DGE 0.1206
8 C 4.6062 -3.2065 0.0069 C.ar 1 DGE -0.0197
9 C 4.2621 -1.8570 0.1708 C.ar 1 DGE -0.0543
10 O 4.1011 -5.3998 -0.7036 O.3 1 DGE -0.4894
11 C 3.1002 -6.2627 -1.2708 C.3 1 DGE 0.1151
12 C3 3.4888 -7.7350 -1.1214 C.3 1 DGE 0.0864
13 O1 4.7488 -7.9743 -1.7458 O.3 1 DGE -0.3887
14 C1 3.5559 -8.1826 0.3300 C.3 1 DGE -0.0357
15 C 2.8918 1.1840 -0.9221 C.ar 1 DGE -0.0372
16 C 3.0523 2.4988 -0.4592 C.ar 1 DGE -0.0543
17 C 3.2536 3.5679 -1.3257 C.ar 1 DGE -0.0197
18 C 3.2961 3.3733 -2.7046 C.ar 1 DGE 0.1206
19 C 3.1537 2.0763 -3.1940 C.ar 1 DGE -0.0197
20 C 2.9490 0.9932 -2.3162 C.ar 1 DGE -0.0543
21 O 3.4565 4.3802 -3.6269 O.3 1 DGE -0.4894
22 C 3.5239 5.7006 -3.0598 C.3 1 DGE 0.1151
23 C4 3.6692 6.7696 -4.1450 C.3 1 DGE 0.0864
24 O2 4.8809 6.5708 -4.8717 O.3 1 DGE -0.3887
25 C2 2.4996 6.7855 -5.1148 C.3 1 DGE -0.0357
26 H 0.5945 -0.7080 0.8319 H 1 DGE 0.0241
27 H 0.4870 -0.0914 -0.8267 H 1 DGE 0.0241
28 H 0.5820 1.0321 0.5106 H 1 DGE 0.0241
29 H 2.7964 -0.4694 2.1962 H 1 DGE 0.0241
30 H 2.4602 1.2314 1.9559 H 1 DGE 0.0241
31 H 4.0596 0.6067 1.5647 H 1 DGE 0.0241
32 H 1.3436 -1.8695 -1.5669 H 1 DGE 0.0622
33 H 1.9409 -4.1449 -1.8761 H 1 DGE 0.0654
34 H 5.5227 -3.5875 0.4515 H 1 DGE 0.0654
35 H 4.9462 -1.2317 0.7348 H 1 DGE 0.0622
36 H 3.0235 -6.0384 -2.3413 H 1 DGE 0.0722
37 H 2.1344 -6.1066 -0.7734 H 1 DGE 0.0722
38 H 2.7427 -8.3440 -1.6445 H 1 DGE 0.0624
39 H 5.3392 -7.2529 -1.4565 H 1 DGE 0.2099
40 H 4.3393 -7.6508 0.8786 H 1 DGE 0.0256
41 H 3.7994 -9.2493 0.3821 H 1 DGE 0.0256
42 H 2.5983 -8.0235 0.8364 H 1 DGE 0.0256
43 H 2.9907 2.7425 0.5967 H 1 DGE 0.0622
44 H 3.3496 4.5491 -0.8758 H 1 DGE 0.0654
45 H 3.1775 1.8944 -4.2680 H 1 DGE 0.0654
46 H 2.8110 0.0055 -2.7501 H 1 DGE 0.0622
47 H 4.4071 5.7580 -2.4113 H 1 DGE 0.0722
48 H 2.6102 5.9083 -2.4888 H 1 DGE 0.0722
49 H 3.7417 7.7485 -3.6575 H 1 DGE 0.0624
50 H 4.9306 5.6173 -5.0723 H 1 DGE 0.2099
51 H 2.4413 5.8552 -5.6885 H 1 DGE 0.0256
52 H 2.6246 7.5977 -5.8389 H 1 DGE 0.0256
53 H 1.5529 6.9377 -4.5866 H 1 DGE 0.0256
@<TRIPOS>BOND
1 1 2 1
2 2 3 1
3 2 4 1
4 4 5 ar
5 5 6 ar
6 6 7 ar
7 7 8 ar
8 8 9 ar
9 4 9 ar
10 7 10 1
11 10 11 1
12 11 12 1
13 12 13 1
14 12 14 1
15 2 15 1
16 15 16 ar
17 16 17 ar
18 17 18 ar
19 18 19 ar
20 19 20 ar
21 15 20 ar
22 18 21 1
23 21 22 1
24 22 23 1
25 23 24 1
26 23 25 1
27 1 26 1
28 1 27 1
29 1 28 1
30 3 29 1
31 3 30 1
32 3 31 1
33 5 32 1
34 6 33 1
35 8 34 1
36 9 35 1
37 11 36 1
38 11 37 1
39 12 38 1
40 13 39 1
41 14 40 1
42 14 41 1
43 14 42 1
44 16 43 1
45 17 44 1
46 19 45 1
47 20 46 1
48 22 47 1
49 22 48 1
50 23 49 1
51 24 50 1
52 25 51 1
53 25 52 1
54 25 53 1
You can see that only C1-C4 are uniquely named. Those unique names will persist forever in HTPolyNet in any system derived from this DGE input file. Other atoms will acquire unique names through processing with AmberTools, but that won’t concern us here.
PACM¶
4,4-diaminodicyclohexylmethane, referred to colloquially as PACM (“pack-em”), is a common hardener in epoxy formulations. Since it has two primary amine groups, it can bond to at most four distinct epoxide groups. The SMILES string for PACM is:
C1CC(CCC1CC2CCC(CC2)N)N
Just as we did with DGEBA, we can generate a structure for the “PAC” monomer:
$ echo "C1CC(CCC1CC2CCC(CC2)N)N" | \
obabel -ismi -h --gen3d -omol2 --title "PAC" | \
sed s/"UNL1 "/"PAC "/ > PAC-raw.mol2
Since we know PACM has two primary amines, we don’t need to convert it to a form with sacrificial H’s – it already has them. We do, however, need to edit PAC-raw.mol2 to give unique atom names to the two amine nitrogens and the two chiral carbons to which they are attached:
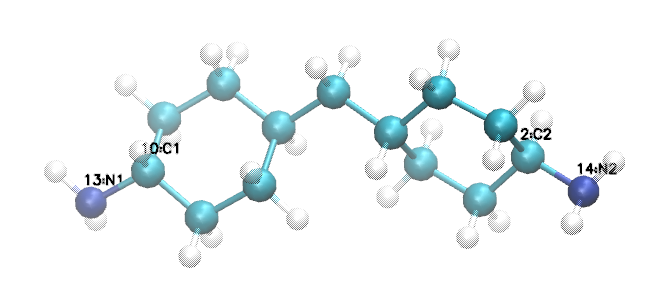
We see that the two amine nitrogens are atoms 13 and 14 in VMD numbering, which correspond respectively to atoms 14 and 15 in mol2 numbering, so let’s call them “N1” and “N2”, respectively. The carbon atom 11 (10 in VMD numbering) to which our “N1” is bound can now be called “C1”, and the carbon atom 3 (2 in VMD) to which our “N2” is bound “C2”.
$ echo PAC-raw.mol2 | sed s/"14 N "/"14 N1"/ | \
sed s/"15 N "/"15 N1"/ | \
sed s/"3 C "/"3 C1"/ | \
sed s/"11 C "/"11 C1"/ > PAC.mol2
Let’s look at the file PAC.mol2 that results from the command above:
@<TRIPOS>MOLECULE
PAC
41 42 0 0 0
SMALL
GASTEIGER
@<TRIPOS>ATOM
1 C 1.0203 1.1686 -0.4045 C.3 1 PAC -0.0488
2 C -0.3868 1.4530 0.1332 C.3 1 PAC -0.0375
3 C2 -0.4239 1.5867 1.6509 C.3 1 PAC 0.0049
4 C 0.2189 0.3673 2.3129 C.3 1 PAC -0.0375
5 C 1.6627 0.1840 1.8377 C.3 1 PAC -0.0488
6 C 1.7559 0.0170 0.3181 C.3 1 PAC -0.0407
7 C 3.2445 -0.0611 -0.1651 C.3 1 PAC -0.0474
8 C 4.0849 -1.2509 0.3999 C.3 1 PAC -0.0407
9 C 5.5341 -1.2664 -0.1535 C.3 1 PAC -0.0488
10 C 6.3098 -2.5522 0.1636 C.3 1 PAC -0.0375
11 C1 5.4974 -3.8029 -0.1700 C.3 1 PAC 0.0049
12 C 4.1937 -3.8000 0.6212 C.3 1 PAC -0.0375
13 C 3.3524 -2.5924 0.2247 C.3 1 PAC -0.0488
14 N1 6.2599 -5.0172 0.1162 N.3 1 PAC -0.3272
15 N2 -1.8168 1.7369 2.0786 N.3 1 PAC -0.3272
16 H 1.6047 2.0898 -0.3424 H 1 PAC 0.0268
17 H 0.9019 0.9202 -1.4627 H 1 PAC 0.0268
18 H -1.0564 0.6483 -0.1927 H 1 PAC 0.0280
19 H -0.7633 2.3773 -0.3343 H 1 PAC 0.0280
20 H 0.1247 2.4885 1.9532 H 1 PAC 0.0458
21 H -0.3534 -0.5388 2.0744 H 1 PAC 0.0280
22 H 0.2067 0.4761 3.4022 H 1 PAC 0.0280
23 H 2.0776 -0.7078 2.3325 H 1 PAC 0.0268
24 H 2.2691 1.0366 2.1678 H 1 PAC 0.0268
25 H 1.2371 -0.9012 0.0434 H 1 PAC 0.0301
26 H 3.7432 0.8605 0.1294 H 1 PAC 0.0271
27 H 3.2593 -0.0975 -1.2596 H 1 PAC 0.0271
28 H 4.1835 -1.0879 1.4730 H 1 PAC 0.0301
29 H 6.0813 -0.4176 0.2686 H 1 PAC 0.0268
30 H 5.5482 -1.1352 -1.2427 H 1 PAC 0.0268
31 H 6.5982 -2.5580 1.2292 H 1 PAC 0.0280
32 H 7.2463 -2.5515 -0.4099 H 1 PAC 0.0280
33 H 5.2588 -3.8065 -1.2451 H 1 PAC 0.0458
34 H 4.3975 -3.7699 1.7048 H 1 PAC 0.0280
35 H 3.6232 -4.7131 0.4347 H 1 PAC 0.0280
36 H 2.4417 -2.5989 0.8401 H 1 PAC 0.0268
37 H 3.0264 -2.7222 -0.8175 H 1 PAC 0.0268
38 H 6.5386 -5.0249 1.0976 H 1 PAC 0.1185
39 H 7.1205 -5.0120 -0.4231 H 1 PAC 0.1185
40 H -2.3522 0.9246 1.7729 H 1 PAC 0.1185
41 H -2.2309 2.5311 1.5900 H 1 PAC 0.1185
@<TRIPOS>BOND
1 1 2 1
2 2 3 1
3 3 4 1
4 4 5 1
5 5 6 1
6 1 6 1
7 6 7 1
8 7 8 1
9 8 9 1
10 9 10 1
11 10 11 1
12 11 12 1
13 12 13 1
14 8 13 1
15 11 14 1
16 3 15 1
17 1 16 1
18 1 17 1
19 2 18 1
20 2 19 1
21 3 20 1
22 4 21 1
23 4 22 1
24 5 23 1
25 5 24 1
26 6 25 1
27 7 26 1
28 7 27 1
29 8 28 1
30 9 29 1
31 9 30 1
32 10 31 1
33 10 32 1
34 11 33 1
35 12 34 1
36 12 35 1
37 13 36 1
38 13 37 1
39 14 38 1
40 14 39 1
41 15 40 1
42 15 41 1
The next thing we consider is how to create the reaction dictionaries necessary to describe the crosslinking chemistry.